Table of Contents
R/qtl interface
This tutorial describes how to connect XGAP to the RqtlPackage.
Retrieve raw data sets from XGAP into R/qtl for analysis
- Connect XGAP to R as described in RprojectInterface.
- Load the R/qtl package by entering (we assume you installed it ;-)).
library(qtl)
- Import an a genotype (individual x marker) matrix and a phenotype matrix (individual x trait) into an R/qtl 'cross' object from XGAP by using the CrossFromMolgenis function. The first parameter denotes the investigation_name, the second and third parameter refer to data matrices. (Tip: list all data matrices available using
find.data()
Here we use again the MetaNetwork set as example.
myCross <- CrossFromMolgenis("MetaNetwork" ,"data_genotypes", "data_metaboliteexpression")
- Perform a standard QTL scan on all traits by using the 'scanall' method which a wrapper around a custom 'scanone' function that handles trait names properly.
myResults <- scanall(myCross)
Pushing results back-and-forth between XGAP and R/qtl
The results can be handled as any R/qtl data.
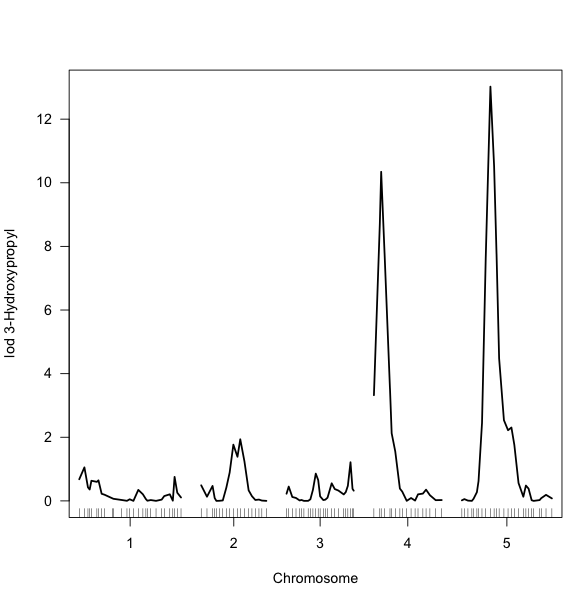
- For example, make a plot of the first trait by selecting the first 'scanone' object from the result list.
plot(myResults[[1]])
Result:

- Push the results back to the XGAP database by using the ResultsToMolgenis function. The name for the datamatrix that will be created may not yet exist in the database and will be automatically created.
ResultsToMolgenis("MetaNetwork", "myResults", myResults)
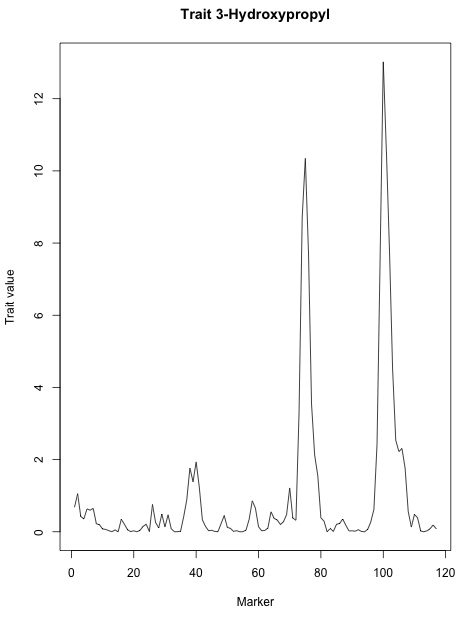
- You can now use the "Rplot" plugin at the Matrices to also visualize this profile, in a generic way:

- You can access past results by using ResultsFromMolgenis?:
myOldResult <- ResultsFromMolgenis("MetaNetwork", "myResults")
- and plot it again:
plot(myOldResult[[1]])
Last modified 16 years ago
Last modified on 2010-10-01T23:38:13+02:00
Attachments (2)
- rqtl-metanetwork-trait1plot.png (27.4 KB) - added by 17 years ago.
- xgap-metanetwork-trait1plot.png (27.2 KB) - added by 17 years ago.
{kind=link}
{kind=link}
Download all attachments as: .zip